

In my previous article, EU MDR Implementation: Important Dates and Challenges, we examined the effects of the new EU MDR on medical device manufacturers. The European Union Medical Device Regulation (EU MDR), which replaced the Medical Device Directive (MDD) in May 2017, has significantly impacted the industry.
Compliance with the EU MDR is essential for marketing medical devices within the EU, as it introduces new requirements to ensure patient access to care. As part of these regulatory changes, EU MDR Article 18, also known as the “Patient Implant Card,” introduces new obligations that specifically address how information about implanted devices is communicated to patients.
In this blog post, we’ll examine EU MDR Article 18 and its specific requirements. We’ll explore what manufacturers need to do to comply with this new regulation, its implications for healthcare institutions, and how these changes will impact the overall delivery of patient care. Understanding Article 18 is crucial for navigating the complexities of EU MDR compliance and ensuring that your devices meet the new standards for patient information.
Table of Contents
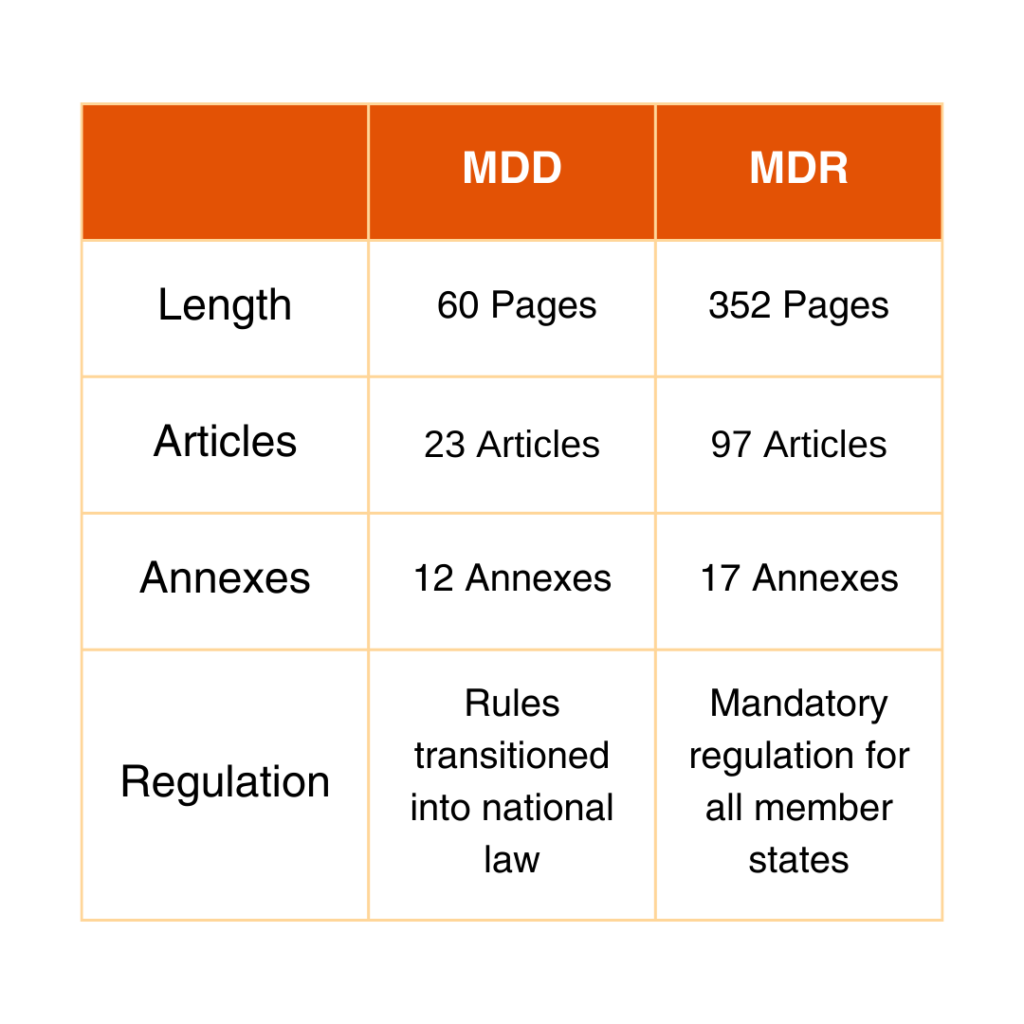
Comparing MDD and MDR
The differences between the MDD and the new MDR are substantial:
Organizations must undertake extensive internal work to prepare for Notified Body (NB) assessments of Technical Documents (TD), requiring a structured approach that involves every business function, from product ideation to distribution.
EU MDR Article 18: The Patient Implant Card
EU MDR Article 18, the “Patient Implant Card,” introduces new obligations for manufacturers regarding patient information.
EU MDR Article 18: Implant Card and Information to be Supplied to the Patient with an Implanted Device
Manufacturer’s Obligations
- Provide information for device identification, including the device name, serial number, lot number, UDI (Unique Device Identification), device model, and manufacturer details.
- Include warnings, precautions, and measures for patients or healthcare professionals regarding potential external influences, medical examinations, or environmental conditions.
- Offer information on the device’s expected lifetime and necessary follow-up.
- Ensure any other information is provided for the patient’s safe use of the device, including specific details from Annex I.
This information must be easily accessible to the patient, written in a layperson-friendly language, and updated as necessary. Updates should be available via the manufacturer’s website.
Health Institutions’ Obligations
Member States must ensure health institutions provide this information to patients with implanted devices and implant cards bearing the patient’s identity.
Exemptions
Certain implants, such as sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips, and connectors, are exempt from these obligations. The Commission can amend this list as needed.
The above product types are classified under the Well-Established Technologies (WET) category.
Impact on Healthcare Delivery
These obligations under Article 18 place additional demands on the content of Technical Documents (TD), the execution of the End-to-End Supply Chain, and healthcare professionals’ day-to-day responsibilities to patients. As previously pointed out, the mandate must be implemented within the latest transition periods revised by the EU parliament and approved on March 15, 2023.

Key Transition Dates
- May 26, 2024: Manufacturers must have a QMS compliant with MDR and have formally applied to a Notified Body.
- September 26, 2024: Written agreement between the Notified Body and the Manufacturer.
- May 26, 2026: All Class III custom-made implantable devices must be MDR certified.
- December 31, 2027: Class III and Class IIb implantable legacy devices (excluding Well Established Technologies-WET) must have MDR certification.
- December 31, 2028: The legacy devices (Class IIb, Class IIa, Class Is, and Class Im) must have MDR certification.
Any devices up-classified under the new regulation requiring Notified Body involvement must be MDR certified. All legacy devices must comply with EU MDR. The clause for the sell-off period ending on May 27, 2025, has been eliminated.
Exemptions and Impacted Product Families
One challenge a manufacturer might face is incorporating the implant card into the existing product packaging design and manufacturing process flow. Modifications could be required to meet Article 18. This requires an assessment of the existing MDD product to determine if any changes to the validated state must be executed.
Devices exempted from these obligations fall under the Well-Established Technologies (WET) classification. However, these new requirements impact product families such as Hernia Meshes, Pacemakers, Knee, Hip, and Shoulder Implants, Breast Implants, Absorbable Adhesion Barriers, and Adjunctive Hemostats.
Key Definitions
| Implant Identification | Information that uniquely identifies the implant, including its name, serial number, lot number, UDI (Unique Device Identifier), and model. |
| Manufacturer Information | The name, address, and website of the implant manufacturer. |
| Device Warnings, Precautions, and Measures | Any warnings, precautions, or measures to be taken by the patient or healthcare professionals regarding electromagnetic interference, medical examinations, or environmental conditions. |
| Expected Lifetime of the Device | Information on the anticipated operational duration of the implant and any necessary follow-up. |
| Any Other Information to Ensure Safe Use | Additional details that help ensure the patient can use the implant safely, such as MRI safety information or limitations on physical activities. |
| Information Delivery Format | Additional details that help ensure the patient can use the implant safely, such as MRI safety information or limitations on physical activities. |
| Patient Information | Space for the name of the patient who received the implant. |
| Implantation Date | The date when the implant was inserted into the patient. |
| Healthcare Provider Information | Identification of the healthcare institution or provider who performed the implantation. |
| Accessibility | The information must be made available to the patient who has been implanted with the device by means allowing rapid access to information. |
The regulation mandates that patient information be accessible via the manufacturer’s website in addition to the required add-ons to each product unit. Healthcare professionals will also receive an implant card guide to help them navigate their responsibilities to patients.
As you can see, Article 18 introduces several new requirements. Implementing these will necessitate a detailed plan while concurrently addressing the future state through the product family Technical Document (TD) submission.
Compliance Architects LLC can partner with your organization to implement this crucial regulation, ensuring the availability of your products in the EU.
Contact Us
To learn more about EU MDR Article 18, fill out the form below to contact us.
FAQs
What is the European Medical Device Regulation (EU MDR)?
The EU Medical Device Regulation (EU MDR) is a comprehensive regulatory framework established by the European Union to govern the safety and performance of medical devices marketed within its member states. It replaced the previous Medical Device Directive (MDD) in May 2017 to enhance patient safety and ensure more rigorous oversight of medical devices.
What are the new MDR requirements?
The new EU MDR requirements include:
Patient Implant Card (Article 18): Manufacturers must provide detailed information on implanted devices, including device ID, warnings, expected lifetime, and safe use instructions. This must be accessible to patients and available on the manufacturer’s website.
Notified Body Assessments: Manufacturers need extensive internal documentation for Notified Body reviews, with deadlines for MDR certification based on device class.
Compliance Deadlines: Specific dates by which various device classes must be MDR certified for quality management systems, starting from May 26, 2024.
Exemptions: Certain Well-Established Technologies are exempt from some new requirements but may be updated by the Commission.
Updated Packaging: Incorporation of the Patient Implant Card into product packaging may require design changes.
What is the difference between EU MDR and the FDA?
The main differences between the EU Medical Device Regulation (EU MDR) and FDA regulations lie in their regulatory approaches and requirements. The EU MDR emphasizes comprehensive safety and performance criteria with detailed post-market surveillance and requires extensive clinical evaluations. In contrast, the FDA’s system includes premarket notifications (510(k)) for many devices, rigorous premarket approval (PMA) for high-risk devices, and focuses on substantial equivalence. The FDA also has ongoing post-market surveillance but with a different emphasis than the EU MDR’s detailed mandates.
What is the US equivalent of the MDR?
The FDA’s Code of Federal Regulations is similar to the EU MDR.
What countries does MDR apply to?
The EU MDR applies to all EU Member States.



