

The implementation European Union Medical Device Regulation (EU MDR) has had a profound impact on the medical device industry. Compliance with this EU MDR implementation, which replaced the Medical Device Directive (MDD) in May 2017, is essential for selling medical devices in the EU.
Table of Contents

Key Aspects of the EU MDR Implementation
Governing Production and Sale: The regulation oversees the production and sale of medical devices distributed in the EU, and it also affects non-EU countries using CE marking.
Consistent Implementation: Ensures uniform implementation and enforcement across the European Union.
Impact on Product Lifecycle: Influences the entire product lifecycle, from design (R&D) to post-market use and discontinuation.
Enhanced Safety and Transparency: Aims to improve patient safety, ensure high-quality products, and increase transparency.
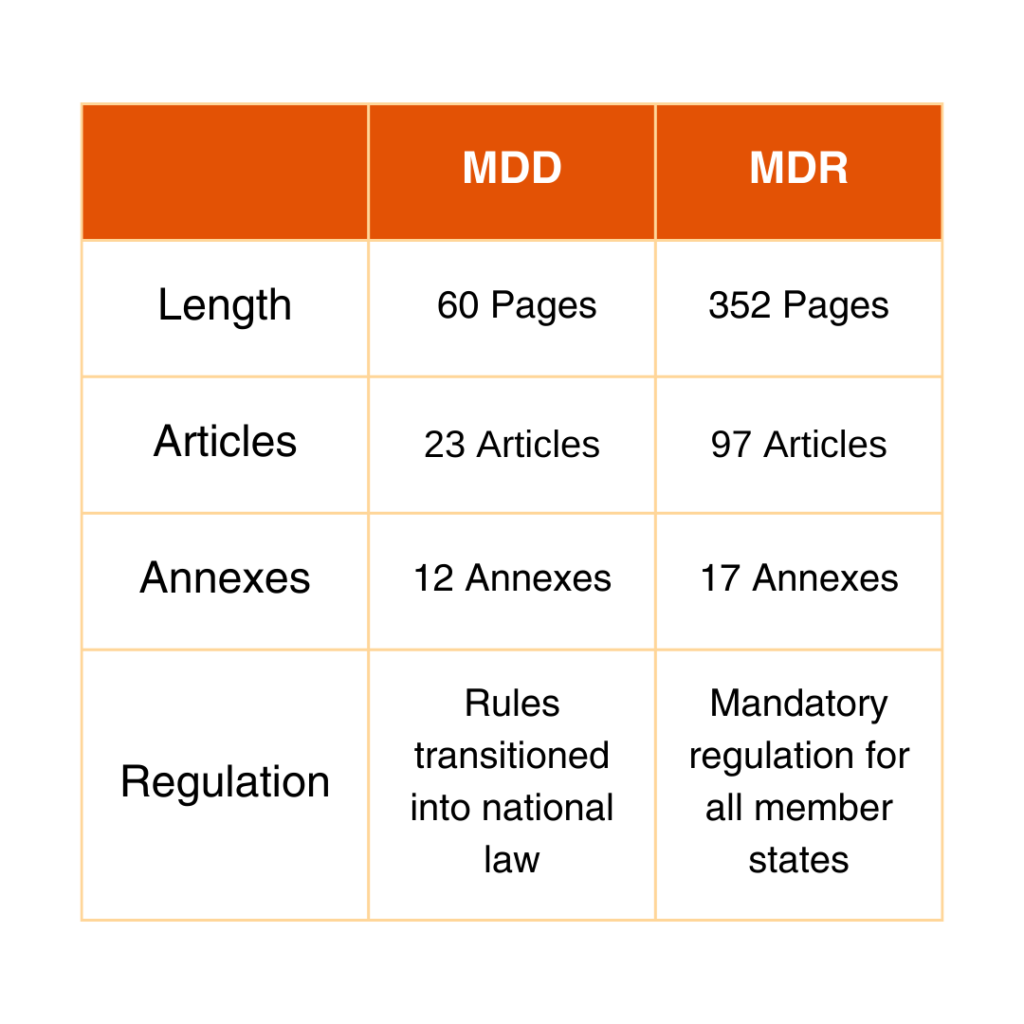
Comparison Between MDD and MDR
See the chart below for the differences between MDD and MDR.
High-Level Requirements in the New Regulation
A. Transparency for Product Safety, Performance, and Use
Market Overview and Product Identification: Introduction of Unique Device Identifier (UDI) and provision for implant cards.
Safe Product Use Information: Instructions for use (IFUs) and implant leaflets in applicable languages.
Public Information on Product Safety: Records for the European Database on Medical Devices Regulation (EUDAMED), including clinical trial data, post-market reports, and safety summaries.
B. Increased Responsibility of Market Actors
Self-Control of Manufacturers: Assignment of a person responsible for Regulatory Compliance. All economic operators (EO) must ensure conformity of marketed products.
Compliance Verification: Downstream EOs (e.g., importers) must conduct compliance verification checks.
Liability of Authorized Representatives: Authorized representatives must have the same liability insurance as the legal manufacturer.
C. Stricter Requirements for Market Access
Recertification of Product Portfolio: Stricter regulation requires recertification, with a higher external review percentage by notified bodies (from ~20% to ~80%).
Extensive Certification Documentation: More extensive information on products, provided in a clear, readily available, and searchable format.
Supervision by Authorities: Stricter supervision by health authorities and notified bodies, including unannounced audits, inspections, and increased vigilance reporting.
Comprehensive Clinical Evidence: Regular (yearly or bi-yearly) clinical evidence updates for high-risk class devices and implantable products.
Revised Transition Periods and Implementation Challenges
The limited capacity of the Notified Bodies (NBs) and the unpreparedness of manufacturers have created significant challenges in implementing the MDR as per the initial May 27, 2025, timeline. This was the end of the selloff period for MDD certified products. Now the revised transition periods (approved on March 15, 2023) are:
May 26, 2024: Manufacturers must have a QMS compliant with MDR and have formally applied to a Notified Body.
September 26, 2024: Written agreement between the Notified Body and the Manufacturer.
May 26, 2026: All Class III custom-made implantable devices must be MDR certified.
December 31, 2027: Class III and Class IIb implantable legacy devices (excluding Well Established Technologies-WET) must have MDR certification.
December 31, 2028: The rest of the legacy devices (Class IIb, Class IIa, Class Is, and Class Im) must have MDR certification.
Any devices up-classified under the new regulation requiring Notified Body involvement need to be MDR certified. All legacy devices must comply with EUMDR.
The clause for the sell-off period ending on May 27, 2025, has been eliminated.
Operational Costs
Transitioning to the EU MDR implementation involves initial costs, potentially including capital investments to meet new requirements such as eIFU databases, patient implant cards, and leaflets. For some medical device organizations, implementation costs can reach up to 5% of top-line sales, influenced by the number of SKUs and device complexity!
Additionally, sustainable activities for maintaining MDR certificates must be factored into future operational costs. These activities will require ongoing surveillance audits and reviews to ensure continued compliance. R&D activities and end-to-end supply chain processes must adopt these new requirements. However, there may be excellent opportunities to streamline portfolios by harmonizing product offerings, thereby leveraging any incremental costs.
Key Milestones
Organizations transitioning their portfolios in the European Union should maintain a tracking dashboard and allocate adequate resources to meet key milestones:
MDR Risk Class
Determine the medical device’s MDR risk class to promptly identify the appropriate transitional timeline, as per the amended MDR regulations.
Quality Management System Remediation
After conducting a gap analysis, the entire QMS and supporting processes must be updated to ensure compliance. Then, the Notified Body (NB) will conduct the corresponding audit.
Gap Assessment
Execute a comprehensive gap assessment for the respective medical device(s) certified under the Medical Device Directive (MDD)/ Active Implantable Medical Devices Directive (AIMDD), identify and address any non-conformances with the MDR regulations, and ensure timely compliance.
Technical Documentation Files (TD)
Existing files must be updated to comply with MDR. The corresponding NB will issue new MDR certificates. The assessment process may require a review by a Competent Authority (CA) in addition to the NB’s conformity assessment.
This review process might take up to twelve months. The new regulation significantly increases the TD content, translating into thousands of pages of information with expanded requirements. Some organizations report MDR TDs containing 50,000 pages!
Communication
To ensure compliance with the MDR regulations, it is crucial to identify and initiate communication with the MDR-designated NBs possessing the specific competency required for classifying medical devices.
Most organizations’ current MDD-notified bodies have been certified under MDR. Otherwise, upgrading to MDR and shifting to a new NB could aggravate the complexity of this journey!
Effective communication with the NB is critical to understand the conformity assessment review process. Ensure that subject matter experts (SMEs) can quickly answer questions and clarify content. The turnaround time with the NB follow-up information/questions is critical to achieving the TD certificate.
A successful transition depends on the construction of the TD and the relationship with the NB.
Conclusion
Time is of the essence! Manufacturers must take prompt action to ensure compliance with the EU MDR implementation. The extended timeline creates opportunities; however, the certification processes are arduous and complex.
Once the MDR certificate is issued, internal product manufacturing conversions are executed. Most organizations do not have the luxury of putting large quantities of inventory at risk, given their supply chains’ end-to-end capacity limitations.
Contact Us
To learn more about the EU MDR implementation, please get in touch with our team by filling out the form below.



