

The FDA and other global regulators rely on data reviews during inspections to determine whether a pharmaceutical or medical device manufacturer operates within regulatory requirements and expectations. The credibility of that data is a crucial parameter in the regulator’s assessment of whether operations are compliant—or not.
Data integrity refers to the credibility—reliability, validity, authenticity, and trustworthiness—of various data presented for regulatory review. Questions about the integrity of data presented in regulator-reviewed documentation always raise red flags about the compliance state of your operation and the levels of control within the medical product manufacturing system.
Understanding Data Integrity
Data integrity encapsulates the reliability, validity, authenticity, and trustworthiness of information submitted for regulatory assessment. Any doubts about data credibility in regulatory documentation trigger concerns about operational compliance and control within the medical product manufacturing ecosystem.
Since data collection and reporting are foundational for cGMP requirements and to support product clearance, approval or licensing, data integrity is foundational to all aspects of regulatory compliance. All regulators, the FDA, and other international bodies take the concept very seriously.
Significance and Regulatory Focus
Questions about data integrity are among those most frequently appearing as FDA Form 483 observations and warning letter citations. Out of 15 cGMP-related warning letters issued to drug firms and four QSR-related letters to device companies in 2019 and posted to the FDA website, approximately nine included at least one citation related to data integrity, according to a review by Compliance Architects®.
The issue was highlighted by a Bloomberg article, which examined problems ongoing at Mylan manufacturing sites, many involving data integrity.
Data integrity deficiencies are not a new trend. In a January 2017 guest column for Pharmaceutical Online, Barbara Unger of Unger Consulting highlighted the emphasis on data integrity in FDA enforcement activity during 2013-2016. The image of this growing emphasis goes back years now.
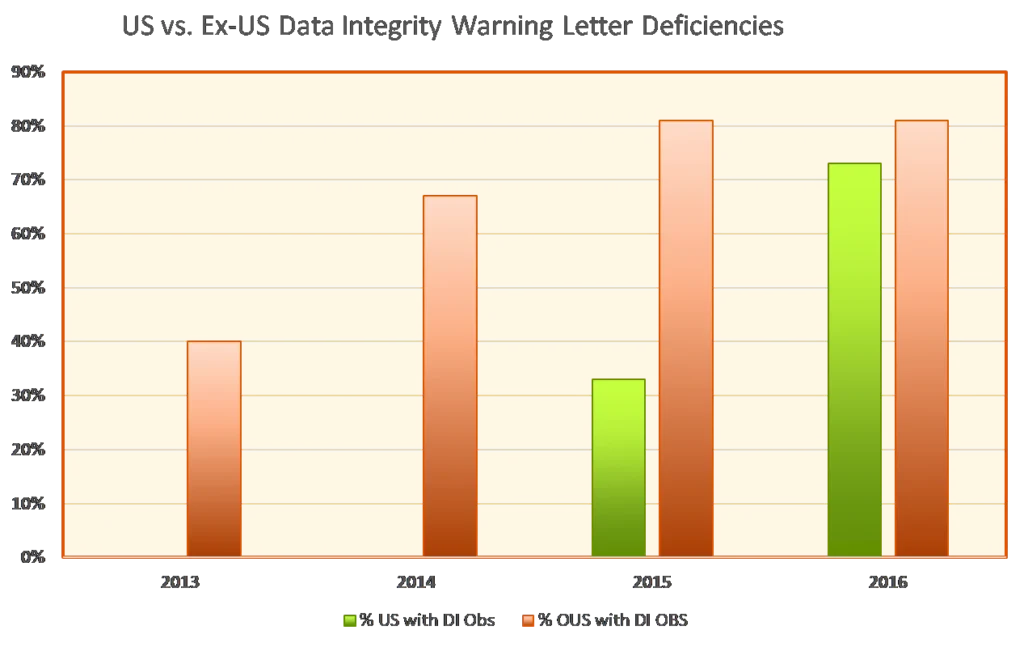
It’s important to note that the words “data integrity” may not appear in such citations. Instead, companies might see such statements as the following:
- Raw materials, intermediates, and finished API analytical results found to be failing specifications or otherwise suspect (e.g., OOT) are retested until acceptable results are obtained. These failing or otherwise equivocal results are not reported.
- Failure to document production and analytical testing activities when they are performed.
- Failure to prevent unauthorized access or changes to data and to provide adequate controls to avoid the omission of data.
- Appropriate controls are not exercised over computers or related systems to ensure that changes in master production and control records or other records are instituted only by authorized personnel.
- Failure to maintain complete data derived from all testing and to ensure compliance with established specifications and standards.
- During our inspection, we observed multiple incomplete, inaccurate, or falsified laboratory records.
Although the wording may vary, all such citations relate to the faith—or lack thereof—that agency investigators have in the information drug and device companies provide about their manufacturing operations. The FDA considers characteristics when determining data credibility, including attributability, legibility, contemporaneous recording, originality, and accuracy, often abbreviated as ALCOA.
Assessment and Key Indicators
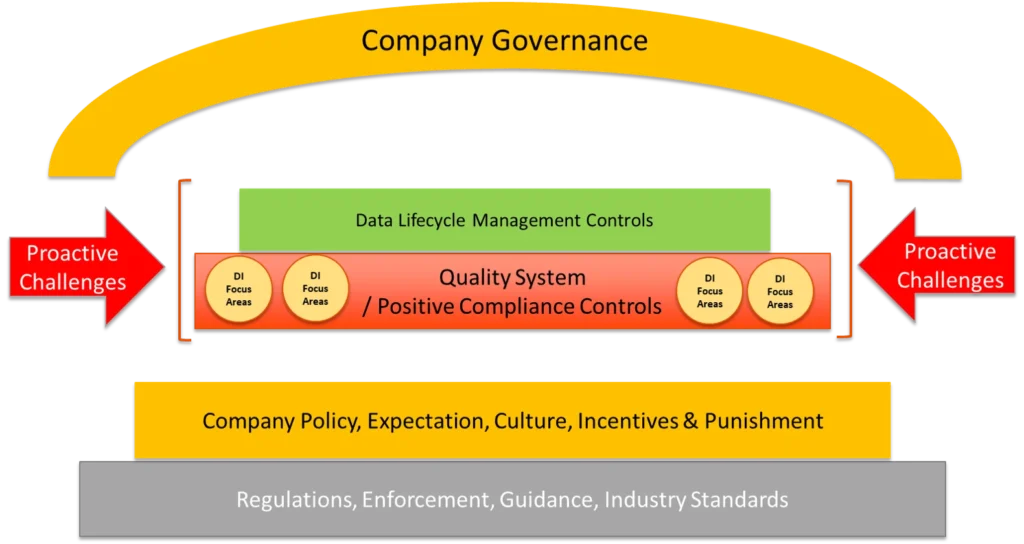
Companies can avoid regulatory and other fallout due to poor data integrity practices by comprehensively implementing a seven-program-element approach within their quality systems (see box below).
Seven Program Elements to Build Data Integrity into Your Systems
- Regulations, enforcement, guidance, and industry standards.
- Policy, expectations, culture, incentives, and punishment.
- Quality system and positive compliance controls.
- Data integrity focus areas.
- Data lifecycle management controls.
- Proactive challenges
- Governance
To fully understand the concept of data integrity, medical product manufacturers need a clear picture of what comprises data from the FDA and other regulators’ perspectives. Examples of data include charts that record information, paper and electronic lab notebooks, product release and approval information, batch release documentation, certificates of analysis, raw data, instrument printouts, and computer-based data. Data includes data stemming from laboratory and clinical studies, and any data supporting a regulatory submission in addition to any data generated in support of cGMP compliance.
Addressing Data Integrity Challenges
Companies can face numerous obstacles to having reliable data and information to present for inspections or regulatory review. Many challenges come down to unintentional errors or negligent conduct due to:
- Lack of awareness;
- Lack of defined expectations from management;
- Lack of procedural or positive controls;
- Lack of adequate supervision and oversight;
- Failure to prioritize the importance of data integrity;
- Tolerance for sloppy or unprofessional work;
- Lack of periodic “checks” on performance;
- Lack of technology controls;
- Pressure on personnel to achieve outcomes; and
- A “whatever it takes” culture.
Data integrity gaps may sometimes be due to purposeful, deliberate conduct to gain monetary, professional, or personal gain. This can occur when the risk of non-compliance is more significant than wrongful conduct. Suppose management creates a culture that presents data manipulation as a victimless crime or directs staff to misrepresent numbers to meet customer service requirements or for financial gain. In that case, employees may fail to ensure the integrity of data presented in FDA-facing records reviewed during inspections.
However, the FDA and other regulators don’t care whether data integrity gaps are due to deliberate or unintentional acts. As far as the FDA is concerned, if it cannot rely on the data, it cannot be assured that manufacturing operations are compliant. And the ramifications for non-compliance can be severe—both in enforcement and customer perception terms—for pharma and medical device manufacturers.
A Holistic Approach to Data Integrity
As with other cGMP requirements, the FDA expects medical product manufacturers to apply a risk-based approach to data integrity. The overall data integrity compliance risk profile is the sum of all data integrity gaps, which can be grouped into three categories: known and documented gaps, known and undocumented gaps, and unknown, undocumented gaps.
Pharma and medical device manufacturers are responsible for identifying, documenting, and addressing all gaps. Thus, the first step toward ensuring data integrity across all quality-related systems and activities is a comprehensive assessment of all potential gaps so that all areas of concern are identified (known) and documented.
Using the comprehensive seven-program-element approach, companies can be confident that this structural approach will maximize the prospect of ensuring data integrity throughout all operations.
The image below illustrates how several of the seven program elements, which Compliance Architects® will explore in greater depth in future articles, can work together to create a comprehensive data integrity program that produces data in which the FDA and other regulators will have faith.
Are you looking for help regarding Data Integrity? Contact us using the form below.



